Genişletilmiş Taşıyıcılık Testleri
IVF ÖNCESİ TAŞIYICI TARAMA TESTLERİ
Avrupa ve diğer gelişmiş ülkelerdeki tanımıyla nadir hastalıklar; “2000'de 1 ya da daha az sıklıkta görülen, çoğu ilerleyici, metabolik, kronik ve bazıları ölümcül olabilen hastalıklardır”. Dünyada bilinen 8000 civarı nadir hastalık bulunmakta olup her hafta literatürde ortalama 5 yeni hastalık tanımlanmaktadır. Türkiye’de nadir hastalık teşhisi konulan 7 milyon kişi dünyada ise 300 milyon kişi bulunmaktadır. Öte yandan, nadir hastalığı olan çocukların %30’u beş yaşını görmeden kaybedilmekteyken %35’i bir yaşa kadar yaşamaktadır1. Bu hastalıkların %90'ının bilinen bir tedavisinin bulunmaması hastalar, yakınları ve sağlık sistemleri için ciddi psikososyal ve ekonomik sorunlara yol açmaktadır1.
Pek çok genetik hastalığın tedavisi ne yazık ki bulunmamakta olup bu grup hastalığın tedavisi için de “Yetim” ilaçlar kullanılmaktadır. “Yetim” ilaçlar, tedavi edilmesi amaçlanan ama çok nadir hastalıklar olduklarından küçük pazar payı sebebiyle sponsorların ürünün araştırma ve geliştirilmesine yatırdıkları sermayeyi amorti etmelerine izin vermeyeceği için normal pazarlama koşullarında sponsorların geliştirme konusunda isteksiz olduğu ilaçlardır. Nadir hastalıkların teşhis süreci hastalar ve aileleri için olduğu kadar sağlık sistemleri için de büyük bir sorundur. Yakın zamanda yapılmış bir çalışmaya göre hasta başına yapılan teşhis masrafları ortalama 3.692 ABD Doları düzeyindeyken, her başarılı teşhisin maliyeti ise ortalama 21.099 ABD doları düzeyindedir. Bu teşhis zorluklarının yanı sıra, nadir hastalıklarının büyük kısmının bilinen bir tedavisinin olmaması nedeniyle bir hastanın teşhis almış olması, tedavi görebileceği ve iyileşebileceği anlamına gelmez. Tedavisi olan 24 nadir hastalığın ekonomik yükü üzerine yapılan yakın tarihli bir çalışmaya göre de hasta başı yıllık maliyet ortalama 266.000 ABD Dolarıdır2.
Gebelik öncesi genetik tarama, gelecek nesillerin, çoğunun bilinen bir tedavisi bulunmayan nadir hastalıklardan korunmasının önemli bir bileşenidir. Nadir hastalıklı doğumların azaltılması ve nihayetinde eradikasyonu amaçlı olarak tarama programları uygulanmakta olup “önleyici” nitelikte evlilik öncesi, gebelik öncesi ve gebelik sırasında yapılan tarama testleri ile “erken teşhis ve tedaviye imkân sağlayıcı” nitelikte yeni doğan tarama testleri yapılmaktadır.
Avrupa İnsan Genetiği Derneği (European Society of Human Genetics -ESHG) kişilerin önündeki alternatif üreme seçeneklerini değerlendirmeye imkân sağlayacağından, prekonsepsiyonel dönemin taşıyıcılık tarama testleri için en ideal dönem olduğunu belirtmektedir3. Ek olarak Amerikan Obstetrisyenler ve Jinekologlar Derneği’nin (ACOG) 2017 yönergesine göre hamile kalmayı düşünen veya zaten hamile olan tüm kadınlara taşıyıcılık taraması hakkında bilgi verilmesi önerilmektedir. Aynı yönergede prekonsepsiyonel dönemde test yaptırmanın daha fazla seçenek sunacağı ve karar vermek için daha fazla zaman sağlayacağı belirtilmiştir4. Taşıyıcı çiftler için aşağıdaki üreme seçenekleri sunulmuş olup İleri jenerasyonlarda hastalık prevalansının azaltılması/eradikasyonunda bu yaklaşım etkin bir rol oynadığı bildirilmektedir.
- PGT
- Prenatal Tanı
- Donor sperm ya da oosit kullanımı
- Riski kabul etme
- Partner değiştirme
Örneğin akrabalık oranının %60’larda olduğu Suudi Arabistan’da genişletilmiş taşıyıcılık taramasını yaptırmayan çiftler Preimplantasyon Genetik Test (PGT) geri ödemesi kapsamından yararlanamamakta. Yine İsrail hükümeti geniş tarama panellerini 1978 yılında halk sağlığı taramaları kapsamında uygulamaya başlanmış, sonraki aşamalarda bulunan hastalık/gen sıklığına bağlı olarak panel içeriğini popülasyona özgü revize etmiştir. Aslında toplumda hastalıkların eradikasyonu konusuna odaklanmış ülkelerin sağlık politikaları kapsamında tarama testi yaparak genetik hastalık taşıyıcılarını belirlemek ve sağlıklı üreme seçeneklerini uygulamak yer almaktadır.
Tarama programları kapsamında uygulanan yenidoğan taraması biyokimyasal testlerle yapılmakta olup patolojik sonuç varlığında tanı ve tedavinin planlanması için yine biyokimyasal testler kullanılmaktadır. Kimi zaman bu testlerin sonuçları arasında tutarsızlıklar olup semikantitatif olan bu değerlerin doğrulanması ve kesin tanı için moleküler testlerin yapılması gerekmektedir. Son yıllarda, genetik analiz için gereken maliyet ve sürenin sürekli azalmasıyla sonuçlanan yüksek verimli dizileme teknolojilerinde kayda değer bir ilerlemeye sebep olmuş ve yeni doğan taramasında yeni nesil dizilemenin (NGS) daha yaygın bir şekilde kullanılmasını sağlamıştır5.
Mümkün olan en yüksek sağlık standardına ulaşma hakkı olarak tanımlanan “sağlık hakkı” temel insan hakkı olan “yaşama hakkı”nın ayrılmaz unsurudur. Sağlık hakkı; uluslararası insan hakları belgeleri, anayasa ve yasalar ile düzenlenmektedir. Bu kapsamda sağlıklı nesillere ulaşmak ve özellikle tedavisi mümkün olmayan genetik hastalıkların önlenmesine yönelik yapılacak çalışmalar daha büyük önem kazanmaktadır.
Ülkemizde Sağlık Bakanlığı tarafından 24 Ekim 2002'de Kalıtsal Kan Hastalıkları Yönetmeliği'nin yayınlanmasının ardından süreç içinde Bakanlığın belirlediği 41 ilde Talaseminin de içinde bulunduğu kalıtsal kan hastalıklarını önleyebilmek için "Hemoglobinopati Kontrol Programı" başlatılmıştır. Program, hükümetin açıkladığı "100 Günlük Eylem Planı" kapsamında, artık 81 ilde "Evlilik Öncesi Hemoglobinopati Tarama Programı" olarak aile hekimleri tarafından uygulanmaktadır. Talasemi hastalığının dünya üzerinde görülme sıklığı 1/10bin iken ülkemizde 1/6bin olarak bildirilmiştir. Yine SMA hastalığının da ülkemizde yaygın görülen (Dünyada 1/10bin – Türkiye 1/6bin) bir hastalık olması sebebiyle Sağlık Bakanlığı 2021 yılı aralık ayı sonu itibari ile 81 ilde SMA Taşıyıcıcılık Taramasını evlilik öncesi sağlık raporu almak için başvuran çiftler ve halen evli olan çiftlerden de talep edenler için uygulamaya başlamıştır. Mevcut uygulamada öncelikle erkek eşten örnek alınmakta ve şüpheli pozitif bir bulgusu olmadığı sürece kadın eşe test yapılmamaktadır6.
Taşıyıcılık teslerinin sonuçlarını etkileyen genomun zorlu gen bölgeleri nelerdir?
Genom üzerinde ortaya çıkan farklı mutasyon türlerinin belirlenmesi amacıyla farklı moleküler yöntemler kullanılmaktadır. Kistik Fibrozis hastalığından sorumlu CFTR geninde genellikle nokta mutasyonlar görülmekte olup dizileme yöntemleri bu tip mutasyonları etkin bir biçimde taramaktadır. Ancak DMD, SMA gibi hastalıklarda delesyon/duplikasyon tipi mutasyonlar yaygın olarak görülmekte olup ancak MLPA (Multiplex ligation-dependent probe amplification) gibi moleküler yöntemlerle bu tip mutasyonlar tespit edilebilmektedir. Genişletilmiş taşıyıcılık panellerinde çok sayıda gen üzerindeki mutasyonları farklı metotlarla tespit etmeye çalışmak laboratuvara ciddi bir iş yükü getirdiği gibi maliyet açısından da testin yaygınlaşmasını sağlayacak etkin yaklaşımı sınırlandırmaktadır. Günümüz teknolojisinde tek bir test ile farklı genlerdeki farklı mutasyon türlerini tespit etmek araştırmacılar tarafından benimsenen bir hedef haline gelmiştir. NGS bu kapsamda en önemli aday test olup biyoinformatik yaklaşımlarla da bu zorlu bölgeleri tespit etme algoritmaları geliştirilmeye devam etmektedir.
Spinal Muscular Atrofi ve Zorlu SMN1 Geni
SMN1 geninin her iki kopyasında defekt olması SMA hastalığına yol açar. SMA hastalığına yol açan mutasyonların çoğu delesyon ya da gen konversiyonlarıdır. SMA hastalığı taşıyıcısı çiftleri tespit etmede bazı komplikasyonlar bulunmaktadır. 5. Kromozom üzerinde yer alan SMN1 ve SMN2 genleri neredeyse tamamen aynı ikişer kopyaya sahip olup iki gen arasındaki başlıca fark ekzon 7 bölgesinin başında yer alan tek nükleotiddir. SMA hastalığı olan bireylerin %98’inde her iki aleldeki SMN1 geninde de anormallik vardır. Bu anormallik homozigot exon 7 delesyonundan (olguların %95’inde) ya da SMN1 genindeki ekzon 7 delesyonu için birleşik heterozigotluktan veya SMN1 geninde delesyon analizleri ile tespit edilemeyen, dizi analizleri ile tespit edilebilen nokta mutasyonlardan kaynaklanır (Şekil 1). Dolayısıyla SMA hastalığı taşıyıcılığı tek bir moleküler test yöntemi ile tespit edilemeyebilmektedir. Taşıyıcılık tanısındaki bir diğer zorluk ise sessiz mutasyonlardır. Bazı kişilerde bir kromozomda iki SMN1 kopyası varken diğer kromozomda SMN1 gen delesyonunun bulunması sessiz mutasyona neden olmaktadır (2+0 SMA taşıyıcılığı durumu). Standart SMA tanı metotları ile tespit edilemeyen sessiz mutasyonları belirlemek taşıyıcılık taramasında hastalığı tam anlamıyla ekarte edebilmek için oldukça önemlidir. Araştırmacılar sessiz mutasyonu tespit edebilmek amacıyla bu mutasyonla birlikte görülen g.27134T>G mutasyonunu taramaktadırlar. Ashkenazi Yahudileri ve Asya toplumları için ilişkili olan bu varyantın diğer etnik popülasyonlardaki tanı etkinliği azalabilmektedir. Bu nedenle sessiz SMA mutasyonları tarama testleri için hala önemli bir test limitasyonudur.
Bir diğer zorluk ise SMA hastalarının %2’si de novo mutasyon taşımakta olup bu mutasyon ebeveynlerinden kalıtılmamakta ve hasta olguda ilk kez ortaya çıkmaktadır7.
Tüm bu sebeplerden ötürü özellikle üreme seçenekleri hakkında danışmanlık almak isteyen çiftlerde SMA taşıyıcılık test sonuçları değerlendirilirken olası tüm tanı limitasyonları göz önünde bulundurulmalı ve gerekirse ek moleküler testlerle riskler azaltılmalıdır.
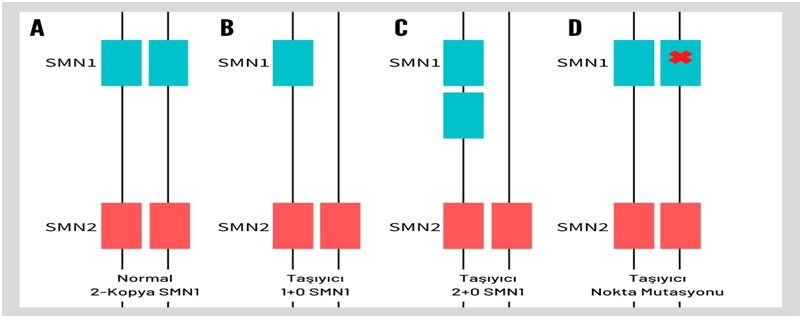
Şekil 1 : (A) Normal – her iki kromozomda 2 kopya SMN1 ve 2 kopya SMN2 (B) SMA taşıyıcısı – bir kromozomda 1 kopya SMN1, diğer kromozomda SMN1 gen kaybı (C) sessiz SMA taşıyıcısı – bir kromozomda SMN1 gen duplikasyonu, diğer kromozomda SMN1 gen kaybı (D) Taşıyıcı - Kromozomun birinde normal SMN1 kopyası, diğer kromozomda SMN1 nokta mutasyonu
Konjenital Adrenal Hiperplazi ve Zorlu CYP21A2 Geni:
Konjenital Adrenal Hiperplazi bir grup resesif hastalık olup 21-Hidroksilaz Eksikliği (21-OHD) en yaygın formudur. Olguların %90-95’inde görülen 21-OHD, CYP21A2 genindeki mutasyonlar nedeniyle ortaya çıkmaktadır. Geniş taşıyıcılık testlerinde gen panelleri içerisinde mutlaka bulunmakla birlikte gen ve psödogeni (CYP21A2 aktif gen, CYP21P psödogen) arasında çok yüksek homoloji olduğundan moleküler tanısı oldukça zorludur. DNA dizisinin intergenik rekombinasyonları sonucunda delesyonlar, duplikasyonlar, gen konversiyonları ve nokta mutasyonlar meydana gelir. CYP21A2’nin psödogeninin bulunması nedeniyle 21-hidroksilaz enzim eksikliğine bağlı gelişen KAH’a sadece CYP21A2 genine yönelik dizi analizi çalışmalarıyla doğru tanı konulmasında çoğu kez aksaklıklar yaşanmaktadır. Ayrıca dizi analizinde saptanması mümkün olmayan kopya sayısı değişikliklerinin belirlenmesi için ek bazı test metodolojilerine de (MLPA) ihtiyaç duyulmaktadır8.
Alfa Talasemi ve Zorlu HBA Geni
Alfa Talasemi, α globulin zincir geninin bir tanesinden dört taneye kadar hepsinin delesyonu veya nokta mutasyonu sonucu, delesyona uğrayan gen miktarına bağlı olarak sessiz anemiden ölümcül hastalık formuna kadar farklı klinik özelliklerdeki bir hastalıktır. α1-globini kodlayan HBA1 ve α2-globini kodlayan HBA2 genleri 16. kromozomun kısa kolunun uç bölgesinde (16p13.3) yerleşik üçer ekzon ve ikişer introndan oluşmaktadır. Etkilenmiş hastaların %90’ında delesyon, %10’unda ise nokta mutasyonları gözlenir. Bu nedenle geniş NGS tabanlı tarama testlerinde bu delesyon tipi mutasyonları tespit etmek oldukça güçtür. (9)
Gaucher Hastalığı ve Zorlu GBA Geni :
Parkinson hastalığına neden olan başlıca risk faktörlerden biri olan Gaucher Hastalığı GBA geninde görülen mutasyonlar sonucu ortaya çıkmaktadır. Son zamanlarda GBA geninin moleküler analizi için tek gen ya da panellerde NGS metodu ile kullanılmaktadır. Ancak Yüksek homolojideki GBAP1 psödogeninin varlığı nedeniyle ortaya çıkan kompleks gen-psödogen etkileşimleri nedeniyle NGS analizi ile mutasyon tespitinde zorluklar oluşmaktadır. Bu nedenle hatalı tanıyı önlemek için dizileme ve analiz yaklaşımlarında bazı stratejiler oluşturulmalıdır. (10)
Kistik Fibrozis ve Zorlu CFTR Geni :
Kistik Fibrozis hastalığı etnik panellerden panetnik panellere taşıyıcılık taramalarındaki en önemli hedef genlerin başında gelmektedir. Her ne kadar CFTR gen mutasyonları çok detaylı incelenmiş ve literatürde birçok çalışma yer almış olsa da her geçen gün özellikle intronik/derin intronik bölgelerde yeni mutasyonlar tanımlanmakta. Sadece kodlayıcı (ekzon) bölgelerin hedeflendiği dizilemelerde bu kodlayıcı olmayan bölge (intron) mutasyonları atlanabilmektedir. Ayrıca delesyon/duplikasyon tipi mutasyonların da varlığı (%3-5) MLPA gibi ek moleküler testlerin yapılmasını gerektirmektedir. Tüm bu mutasyonları tek bir NGS metodu ile yapabilmek taşıyıcılık test panellerinin yaygın ve uygun maliyetli kullanımı için önemli bir hedef haline gelmiştir. (11)
Taşıyıcılık Tarama Programının Tarihçesi:
Genetik taşıyıcılık testleri uzun yıllardır hasta çocuk sahibi olma riski olan genetik hastalık taşıyıcılarının tespit edilebilmesi, bu hastalıkların insidanslarının azaltılması ve taşıyıcı çiftlerin sağlıklı çocuk sahibi olmalarının sağlanmasında önemli bir rol oynamaktadır. Bu tip testlerin kullanımı 1970’lerde Ashkenazi Yahudi popülasyonlarında Tay Sacs hastalığını taramak için kullanılırken 1980’lerde akdeniz ülkelerinde Beta Talasemi ve Orak Hücre Anemisi toplum taramaları başlatıldı. 1993’te aile öyküsü ve etnik kökenine bakılmaksızın Kistik Fibrozis hastalığının toplum taraması programına alınması “Evrensel Taşıyıcılık Testi” tabirini gündeme getirdi. 2010 yılında Consyl isimli biyoteknoloji firması 100 mendeliyen kalıtılan geni içeren bir tarama testini bildirirken 2011 de Bell ve arkadaşları 437 genin tarandığı panelde “Yeni Nesil Dizileme” (Next Generation Sequencing – NGS) teknolojisini kullanan ilk grup olmuştur12. O günden sonra çok hızlı bir biçimde pek çok taşıyıcılık tarama paneli servis edilmiş olup global bir çözüm olarak tüm ekzom / tüm genom tabanlı testlerin kullanımı artmıştır.
Geniş Tarama Testleri (Expanded Carrier Screening - ECS) ilk uygulamaya girdiği dönmelerde “Hot Spot” olarak tabir edilen tüm gen taramaları yerine ilgili genlerdeki yaygın mutasyonları tarayan sınırlı tarama panelleri olarak gündeme gelmişti. Uzun yıllar bu yaklaşımla tarama testleri yapılmış olmakla beraber dizileme teknolojilerindeki gelişmeler ve test maliyetlerindeki iyileşme tüm gen taramalarına imkân verir hale gelmiştir. Bu sayede, intronik ve derin intronik bölgeler gibi genomun patojenik varyant taşıma riski görece daha düşük bölgelerinde de mutasyon analizleri yapılabilir hale gelmiş hastaların nadir taşıyabilecekleri bozukluklar da ekarte edilebilmiştir.
Bu hızlı ve düşük maliyetli testlerle (pan-etnik ya da evrensel) etnik kökene bakılmaksızın eşitlik çerçevesinde ve kişileri damgalamadan taşıyıcılıklarını belirleme şansı kazanılmıştır.
Önceden “Amerikan Kadın Hastalıkları ve Doğum Uzmanları Cemiyeti” (American College of Obstetricians and Gynecologists-ACOG) taşıyıcılık testlerinin etnisiteye göre dizayn edilmesini önermekteydi ve belli hastalıklar için artmış riski olan popülasyonlara odaklanılmaktaydı13.
ACOG 2011 yılında yayınladığı komite kararlarında; taşıyıcılık testlerinin etnik-spesifik, Pan-etnik ve genişletilmiş taşıyıcılık panelleri olarak servis edilebileceğini, evrensel Kistik Fibrozis taramasına SMA ve hemoglobinopati taramasının da eklenmesini önermiştir14. SMA’yı da içeren genetik kondisyonların etnik gruplarla sınırlı olmadığı, globalleşen dünyada etnik grupların karıştığı belirtilmiştir. Bu kapsamda prekonsepsiyonel taramalarda genişletilmiş tarama testlerinin yapılmasının doğru bir strateji olduğu belirtilmiştir. Ancak Kıbrıs, Sardinya Adası gibi Talasemi hastalık insidansı yüksek populasyonlar ve Ashkenazi Yahudi toplumlar dışında taşıyıcılık taramalarının halk sağlığı taramaları kapsamında gebelik planlayan tüm popülasyona uygulanmasının bazı sakıncaları vardır.
- Psikolojik etkiler
- Damgalamak/ayrımcılık
- Kişisel seçimleri usulsüzce baskılaması ve kişisel seçim özgürlüğünü sınırlaması
- Aile kurmak isteyen çiftler arasında seçim kriteri haline gelebilme durumu
Taşıyıcı bireylerin tespitine yönelik testler iki ayrı grupta toplanabilir:
Taşıyıcılık Taraması: Belli bir hastalık için hasta çocuk sahibi olmada artmış riski bulunmayan çiftler
Taşıyıcılık Testi: Eş ya da aile öykülerinde pozitif bulgusu olan hasta çocuk sahibi olma riski artmış çiftler
Varyant Sınıflandırma American College of Medical Genetics – 2010 Genom dizilemesi ile bir bireyde 4 milyon civarında varyant tespit edilirken, genomun protein kodlayan ve %1’lik kısmını oluşturan ekzom dizilemesi sonucunda yaklaşık 20,000 varyant elde edilmektedir. Bu kadar varyanttan hastalık etmeni olan mutasyonu bulmak samanlıkta iğne aramaya benzemektedir. Bu noktada ailenin detaylı klinik bilgisine sahip olmak çok önemlidir (Lohmann ve ark.). Etkin bir biyoinformatik filtreleme için temelde varyantın popülasyon frekansı düşük olanların seçilmesi ile varyant sayısı azaltılabilir ve sonrasında kalıtım paterni ve fenotipik bulguların eklenmesi ile hastalık etmeni mutasyona ulaşılabilir. Klinik tanı net olmadığında kullanılan tüm genom ve ekzom testleri pek çok hasta için yüz güldürücü sonuçlar vermekle birlikte bu yüksek çıktılı engin genom datasının bizlere sunduğu, literatürde daha önceden hiç bildirilmemiş yeni varyantlar şüpheli raporların üretilmesine de neden olmaktadır. Buradaki tehlikeyi anlamak için yeni jenerasyon mutasyon sınıflandırması hakkında fikir sahibi olmamız gerektiğinden dünya üzerinde pek çok klinik raporun dayanağı olan American College of Medical Genetics and Genomics (ACMG) kılavuzu kriterlerine göre mutasyon skorlamasından bahsetmemiz gerekmektedir15 (Şekil 2).
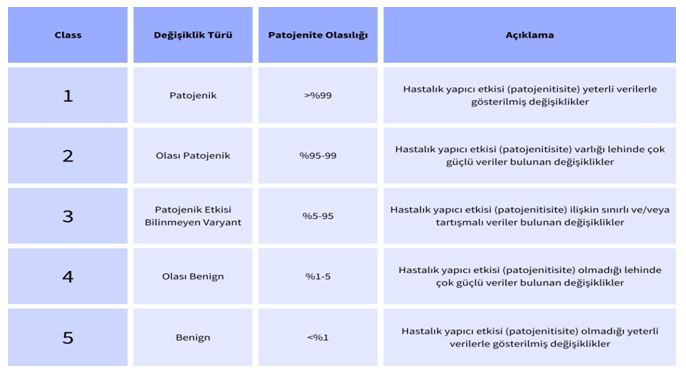
Şekil 2 : American College of Medical Genetics and Genomics (ACMG) kılavuzu kriterlerine göre mutasyon skorlaması
Taşıyıcılık Tarama Testlerinde Hangi Genlere Bakılmaktadır?
Taşıyıcılık taraması erken dönem ölümle seyreden ya da engelli hasta çocuk sahibi olma riski olan taşıyıcı bireylerin tespit edilmesini sağlayan genetik testlerin bütünüdür. Bu testler genellikle resesif ve X’e bağlı aktarılan hastalıkları taramaktadır. Pozitif aile öyküsü olan erişkinlere taşıyıcılık taraması önermek pek çok ülkede standart bir uygulama olmasına rağmen bu yolla taşıyıcı çiftlerin çok az bir kısmı tespit edilebilmektedir. Pek çok hasta çocuk aile öyküsünde hasta birey olmaksızın doğmakta ve sadece yüksek risk kategorisindeki akraba evliliği yapan çiftler tarama testi talep etmektedirler.
Amerikan Tıbbi Genetik ve Genomik Cemiyeti (American College of Medical Genetics and Genomics -ACMG) Ashkenazi Yahudilerin taranması için ilkin 9 gen içeren bir pan-etnik panelin kullanılmasını önermiştir.3 ACOG 2017 yılında yayınladığı komite raporunda 14 genin taranmasını tavsiye etmiştir4. Genel olarak SMA, Tay Sacs, Beta Talasemi, Kistik Fibrozis, Frajil X hastalıklarının tarandığı etnik paneller yaygın olarak kullanılmaktayken dizileme teknolojilerindeki gelişmeler ve test maliyetlerindeki azalma 100-600 gen içerikli panetnik panellerin de yaygınlaşmasına neden olmuştur. Ancak bu testlerde gen panellerinin içeriğinin belirlenmesi hala tartışma konusudur. Üreme tıbbında kullanılmak üzere kişinin etnik kökeni ve aile geçmişi bilgisine gerek duyulmaksızın uygulanabilecek panetnik ve yüksek klinik fayda sağlayan taşıyıcılık testlerinin kullanımı her geçen artmaktadır. Her toplumun kendi epidemiyolojisine uyumlu, farklı ECS yaklaşımlarına ihtiyacı vardır.
Popülasyon genetik çeşitliliği, çevresel, toplumsal faktörler ve hayatta kalma oranları nedeniyle coğrafi bölgeler arasında sıklık bakımından değişiklik gösteren 1800'den fazla kalıtsal nadir hastalık olduğu tahmin edilmektedir16. Avrupa'da sadece 12 nadir hastalığın tüm vakaların %90'ını oluşturduğu bilinmektedir. Bununla birlikte Türkiye ve Orta Doğu ülkelerinde aynı 12 hastalığın gözlenme oranı %35'tir17. Dolayısıyla ülkemizde dünyada sık görülen hastalıklara nazaran oldukça nadir görülen genetik hastalıklar daha yaygındır. Bu durumda evlilik öncesi tarama programlarını genişletmek ve ülkenin epidemiyolojisine uyumlu daha fazla hastalığı tarayan programlara geçmek bu hastalıkların eradikasyonunda büyük önem taşımaktadır. Akraba evliliği oranlarının yüksek olduğu toplumlarda, nadir heterozigot (asemptomatik) taşıyıcılıkların, çocuklarda homozigot olarak (hasta) ortaya çıkma olasılığı artmakta ve bebeklerin bilinen nadir hastalıklardan veya yeni sendromlardan etkilenme riski yükselmektedir.
Göç dalgaları kaynaklı gen havuzumuzun değişmesi ile birlikte yeni genetik hastalıklar ortaya çıkabilmektedir. Özellikle ülkemizde göç eden ülkelerin de akraba evliliği oranının yüksek olduğu göz önünde bulundurulursa genetik hastalıklı çocuk doğumlarının artışı önemli bir sağlık problemini de beraberinde getirmektedir. Benzer şekilde Türkiye'de 3,6 milyon kayıtlı Suriyeli, diğer milletlerden (Irak, Afgan, diğerleri) de 320.000 mülteci yaşamaktadır18. Türkiye İstatistik Kurumu, Suriyeli mültecilerin %30'unun üreme çağında olduğunu, evliliklerin yaklaşık %40'ının akraba evliliği olduğunu ve son dört yılda 518.730 doğum gerçekleştiğini bildirmektedir. Sürekli göç, mülteci topluluklarındaki yüksek üreme ve akraba evliliği oranları nedeniyle, nadir hastalıklarla mücadele Türkiye için öncelikli bir halk sağlığı problemi olarak yeni bir boyut kazanmıştır. Yaygın nadir hastalıkların henüz bilinmeyen genetik varyantları ve yeni sendromlar Türkiye ve Avrupa'nın genetik yapısına dahil olmakta ve kısa vadede bunların görülme sıklıklarının artması beklenmektedir.
ECS, sağlıklı ebeveyn adaylarına gebelik öncesi genetik hastalık taşıyıcılık risklerinin tespit edebilmek amacıyla yapılan, klasik genetik taramaların aksine, yüzlerce hedef gen bölgesinin tek bir testte taranmasını sağlayan, uygulanmasında kişilerin etnik ya da sağlık durumuyla ilgili herhangi bir kriter aranmayan bir genetik taramadır. Bu taramanın yapılması ve nadir hastalık taşıyıcılık durumlarının raporlanması, ebeveyn adaylarının üreme seçenekleri hakkında bilgilendirilmesi ve sağlıklı bebeklerin doğumu için çok önemlidir.
TÜİK aile yapısı araştırması 2016 verilerine göre; Türkiye’de ortalama akraba evliliği oranı %20.9 olup Diyarbakır’da %37.2 Konya’da %23.3’tür. Akraba evliliklerinde çiftler özellikle nadir resesif hastalıkları taşıdıklarından bu hastalıkların pek çoğu genişletilmiş tarama panelleri kapsamında yer almadığından bu tür ticari rutin testler akraba evliliği yapan çiftler ya da akraba evliliğinin yüksek oranda görüldüğü ülkelerin tarama programları için yeterli gelmemektedir. Şimdilerde tarama programları içine girmeye başlayan tüm ekzom (Whole Exome Sequencing-WES) ve tüm genom (Whole Genome Sequencing-WGS) dizilemeleri genetik olarak izole popülasyonların nadir hastalıklarını da içeren tüm hastalıkların taranmasına imkân sağlayacaktır. Ancak yeni nesil dizileme teknolojileri ile elde edilen bu büyük varyant datasının en tehlikeli çıktısı olan bildirilmemiş değişimlerin yorumlanması bu testlerin sonuçlarını kompleks hale getirmektedir. Klinik fenotipe etkisi bilinmeyen, “VUS- Variant of Unsignificance” olarak tabir edilen bu varyantlar patojenik ya da beningn olabilirler. ClinVar gibi halka açık veri tabanlarında varyantların laboratuvarlar arası patojenite değerlendirmeleri arasındaki farklılıklar da sonuçların yorumlanmasında zorlukları beraberinde getirmekte. Bir laboratuvarın patojenik dediği bir varyanta başka bir laboratuvar VUS diyebiliyor. Bunların ötesinde farklı laboratuvarlar aynı kişi için birbirinden tamamen farklı sonuçlar verebiliyor. Genomik alanda teknoloji hızla ilerlerken verileri yorumlamada aynı hızı gösterememekte olduğumuzdan patojenik etkisi net olmayan varyantların genetik danışmanlığının verilmesinde detaylı hasta bilgilendirmesinin yapılması ve hasta onamı ile prenatal ve preimplantasyon testlerin gerçekleştirilmesi büyük önem taşımaktadır.
Panel içeriği genişledikçe bu testlerde duyarlılık sorunları nedeniyle klinik fayda ve uygulanabilirlik yönünden iyileştirmelere ihtiyaç bulunmaktadır. Buna ek olarak bir ECS panel içeriğinin ne olması gerektiği konusunda bir fikir birliği bulunmamaktadır. Bazı ECS testleri yukarıda belirtilen sorunu panele dahil edilen gen sayısının arttırılması ile çözmeye çalışmaktadır. Ancak hem kişisel sağlık hem de halk sağlığı açısından, panelde yer alan gen sayısının artırılması, çiftler arasındaki kümülatif mutasyonların tespiti üzerinde sınırlı bir etkiye sahiptir yani tespit edilen mutasyon sayısı, paneldeki gen sayısı arttıkça iyileşmemektedir. Bu durum panel gen listesi kürasyonunun öneminin altını çizmektedir.
Günümüzde yaygın görüş özgüllük, duyarlılık ve dengeli okuma derinliğinin panelde yer alan gen sayısından daha yüksek önem taşıdığı, 100 hastalığı %100 duyarlılıkla tarayan bir taşıyıcı testinin, 1000 hastalığı %10 duyarlılıkla tarayan tarama testinden daha etkili olduğu yönündedir20.
Genetik veri tabanları, nadir hastalıkların varyantları ve bunların frekansları hakkında bilgi sağlamaktadır, ancak belirli etnik gruplar, yaş grupları ve cinsiyetlere dair veriler yetersiz kalmaktadır. Bu nedenle, 1.000'den fazla monogenik nadir hastalıktan hangilerinin, hangi genlerin ve bunlarla ilişkili hangi varyantların bir ECS paneline dahil edilmesi gerektiği hala önemli bir tartışma konusudur. ECS panelleri, varyantların tespitinde farklı teknik zorluklar barındıran hem kodlayan hem de kodlamayan bölgeleri kapsayan bir gen koleksiyonu içermektedir. Gen panelinin tasarımında, hangi genlerin dahil edileceğine karar verilirken, hedef gen bölgelerini zenginleştirme yönteminin seçimi, okuma derinliğinin belirlenmesi ve panele özgü analiz yazılımı adaptasyonu gibi özellikler açısından kapsayıcı ama aynı anda da hedefe yönelik ve odaklı olmak gerekmektedir21. Genomdaki karmaşık bölgelerdeki varyantların saptanmasında birden fazla akış hattı ve analiz yöntemi kullanılması gerekmektedir. Teknik olarak zorlayıcı varyant türleri için hem algoritma tasarımı hem de yeni verilerle model geliştirilmesi yaklaşımı ile, yapay zekâ tabanlı biyoinformatik araçların kullanımı oldukça önemli bir hale gelmiştir.
Taşıyıcılık testlerinde temel hedef, baştan sona tüm süreci kapsayan bir NGS testi sunmak ve hedef listesindeki DMD, SMA, CFTR gibi zorlu bölgeler için ek test gereksinimini ortadan kaldırmaktır. Klinik düzeyde duyarlılığın sağlanamadığı zorlu genom bölgeleri (psödogenler, gen konversiyonları vb) ve kopya sayısı değişiklikleri (Copy Number Variation – CNV) tespiti için ek bazı mutasyon türüne özgü moleküler test metotları kullanma ihtimali bulunmaktadır.
Varyant sınıflandırması statik bir olgu değildir; Bir varyantın sınıf I, II ya da III olarak sınıflandırılması, kullanılan veri tabanında biriken genetik veriye göre değişebilir. Ayrıca bir varyantın sınıflandırılması farklı veri tabanları arasında farklılık gösterebilir ve bu durum özellikle VUS'ların raporlanmasında sorun oluşturabilir. VUS'ların doğru ve klinik faydayı arttıracak şekilde raporlanması rezidüel (tarama sonuçları herhangi bir taşıyıcılık işaret etmediği durumlarda bile ebeveynlerin taşıyıcı olma olasılığı) riskin azaltılması için büyük önem taşımakta bununla birlikte bu sorumluluk mevcut testler tarafından göz ardı edilerek klinisyene bırakılmaktadır.
Mevcut ECS analizlerinde bir yaklaşım yalnızca patojenik/muhtemel patojenik varyantları (sınıf I ve II) raporlayarak VUS'lar dikkate alınmamakta ve bu da risk altındaki çiftlerin gözden kaçmasına neden olabilmektedir. Eşlerden birinin sınıf I/II varyant taşıyıcısı ve diğerinin ise kodlayan VUS taşıyıcısı olduğu çiftlerin %20'sinin, frekans verilerinin birikimine bağlı olarak, her ikisi de sınıf I/II taşıyıcısı olacak şekilde yeniden sınıflandırılacağı tahmin edilmektedir. Mevcut VUS anlayışı, ECS'de VUS raporlamasını engellese de, bu bulgular VUS'ların sınıflandırmasının ve raporlanmasının önemini vurgulamaktadır. Klinisyene hastalarına doğru üreme tercihleri sunabilmesini sağlayacak, filtrelenmiş VUS bilgisini de içeren bir tarama raporu sağlayabilmek için bulguların gözden geçirilmesi ve karanlıkta kalan noktaların en aza indirilmesi büyük önem taşımaktadır.
ESHG - Genetik tarama programlarının prensipleri, teknikler, uygulamalar, politikalar3.
- Genetik tarama amaçları ve hedef popülasyon iyi tanımlanmalı
- Laboratuvar kalite kontrolleri sıkı olmalı
- Sonuçların limitleri açıkça belirtilmeli
- Otoriteler kişilerin taşıyıcılık durumlarına ait gizliliği sağlayabilmeli ve kişisel genomik verilerin saklanması ve korunmasını güvence altına almalı
- Kişi ve aile mahremiyetini sağlayabilmeli
- Uzun vadedeki sonuçlar monitörize edilebilmeli
- Eğitim programları düzenlenmeli ve genetik danışmanlık verilmeli
Taşıyıcılık testleri kişilerin üreme kararlarının daha güvenilir olmasını sağlamaktadır. Günümüzde geniş taşıyıcılık tarama panellerinin maliyet etkinliği arttığından özellikle IVF uygulamaları öncesi çiftlerin genetik taşıyıcılık risklerinin belirlenmesinde önemli bir uygulama haline gelmektedir. 3877 olgunun dahil edildiği IVF öncesi yapılan ECS test bulgularına göre olguların %10.4’ü en az 1 gende patojenik ya da olası patojenik varyant taşımaktadırlar. Bu çalımada incelenen 766 çiftin %22.6’sında tek eş mutasyon taşıyıcılığı bulunmuşken %2.6’sı risk altındaki her iki eş taşıyıcıları olarak tespit edilmiştir22. Bu kapsamda IVF öncesi taşıyıcılık panellerinin kullanımı hasta çocuk doğumlarının önlenmesinde önemli bir uygulama haline gelmeye başlamaktadır. Ancak özellikle akraba evliliği yüksek toplumlarda sınırlı gen panellerinin yerine daha geniş taşıyıcılık test panellerinin devreye girmesi amaçlanmaktadır. Bu noktada elde edilen genomik verinin etkin değerlendirmesine imkân sağlayacak olan hastalık yapıcı varyant bilgisinin artması hedeflenmektedir. Preimplantasyon Genetik Testlerin günümüz teknolojisinde başarıyla uygulanıyor olması taşıyıcılık testleri ile risk taşıyan çiftler için önemli bir seçenek oluşturmaktadır. Hastalar için taşıyıcılıktan tanıya bir tespit imkânı veren bu üreme seçeneklerinin kullanımı her geçen gün artmaktadır.
Kaynaklar
- https://globalgenes.org/rare-diseases-facts-statistics/
- Andreu P, Karam J, Child C, Chiesi G, Cioffi G. The Burden of Rare Diseases: An Economic Evaluation. :7.
- Henneman, L., Borry, P., Chokoshvili, D. et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet 24, e1–e12 (2016). https://doi.org/10.1038/ejhg.2015.271
- Committee Opinion. (2017b). Committee Opinion No 691 Summary. Obstetrics & Gynecology, 129(3), 597–599. https://doi.org/10.1097/ aog.0000000000001948
- Remec ZI, Trebusak Podkrajsek K, Repic Lampret B, Kovac J, Groselj U, Tesovnik T, Battelino T, Debeljak M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front Genet. 2021 May 26;12:662254. doi: 10.3389/fgene.2021.662254. PMID: 34122514; PMCID: PMC8188483
- https://hsgm.saglik.gov.tr/tr/cocukergen-tp-
- Wirth B, Schmidt T, Hahnen E, Rudnik-Schöneborn S, Krawczak M, Müller-Myhsok B, Schönling J, Zerres K. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet. 1997 Nov;61(5):1102-11. doi: 10.1086/301608. PMID: 9345102; PMCID: PMC1716038.
- Nan MN, Roig R, Martínez S, Rives J, Urgell E, Espinós JJ, Tirado M, Carreras G, Aulinas A, Webb SM, Corcoy R, Blanco-Vaca F, Tondo M. Comprehensive Genetic Testing of CYP21A2: A Retrospective Analysis in Patients with Suspected Congenital Adrenal Hyperplasia. J Clin Med. 2021 Mar 12;10(6):1183. doi: 10.3390/jcm10061183. PMID: 33809035; PMCID: PMC8001222.ALFA
- Vijian D, Wan Ab Rahman WS, Ponnuraj KT, Zulkafli Z, Mohd Noor NH. Molecular Detection of Alpha Thalassemia: A Review of Prevalent Techniques. Medeni Med J. 2021;36(3):257-269. doi: 10.5222/MMJ.2021.14603. Epub 2021 Sep 30. PMID: 34915685; PMCID: PMC8565582.
- Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J Mol Diagn. 2017 Sep;19(5):733-741. doi: 10.1016/j.jmoldx.2017.05.005. Epub 2017 Jul 18. PMID: 28727984.
- Morris-Rosendahl DJ, Edwards M, McDonnell MJ, John S, Alton EWFW, Davies JC, Simmonds NJ. Whole-Gene Sequencing of CFTR Reveals a High Prevalence of the Intronic Variant c.3874-4522A>G in Cystic Fibrosis. Am J Respir Crit Care Med. 2020 Jun 1;201(11):1438-1441. doi: 10.1164/rccm.201908-1541LE. PMID: 32017858.
- Beaudet AL (2015) Global genetic carrier testing: a vision for the future. Genome Med 7:79
- Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. ACOG Committee Opinion No. 442. American College of Obstetricians and Gynecologists. Obstet Gynecol 2009;114:950–3.
- Committee Opinion No. 486: Update on Carrier Screening for Cystic Fibrosis. Obstetrics & Gynecology: April 2011 - Volume 117 - Issue 4 - p 1028-1031 doi: 10.1097/AOG.0b013e31821922c2
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
- Cornel MC, Rigter T, Jansen ME, Henneman L. Neonatal and carrier screening for rare diseases: how innovation challenges screening criteria worldwide. J Community Genet. 2021;12(2):257-265. doi:1007/s12687-020-00488-y
- Beauchamp KA, Muzzey D, Wong KK, et al. Systematic design and comparison of expanded carrier screening panels. Genetics in Medicine. 2018;20(1):55-63. doi:1038/gim.2017.69Refugees and Asylum Seekers in Turkey – UNHCR Türkiye. Accessed August 11, 2022. https://www.unhcr.org/tr/en/refugees-and-asylum-seekers-in-turkey
- Refugees and Asylum Seekers in Turkey – UNHCR Türkiye. Accessed August 11, 2022. https://www.unhcr.org/tr/en/refugees-and-asylum-seekers-in-turkey
- Ben-Shachar R, Svenson A, Goldberg JD, Muzzey D. A data-driven evaluation of the size and content of expanded carrier screening panels. Genetics in Medicine. 2019;21(9):1931-1939. doi:1038/s41436-019-0466-5
- Hogan GJ, Vysotskaia VS, Beauchamp KA, Seisenberger S, Grauman PV, Haas KR, Hong SH, Jeon D, Kash S, Lai HH, Melroy LM, Theilmann MR, Chu CS, Iori K, Maguire JR, Evans EA, Haque IS, Mar-Heyming R, Kang HP, Muzzey D. Validation of an Expanded Carrier Screen that Optimizes Sensitivity via Full-Exon Sequencing and Panel-wide Copy Number Variant Identification. Clin Chem. 2018 Jul;64(7):1063-1073. doi: 10.1373/clinchem.2018.286823. Epub 2018 May 14. PMID: 29760218.
- Liu Z, Zhu L, Roberts R, Tong W. Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends in Genetics. 2019;35(11):852-867. doi:1016/j.tig.2019.08.006
- Capalbo A, Fabiani M, Caroselli S, Poli M, Girardi L, Patassini C, Favero F, Cimadomo D, Vaiarelli A, Simon C, Rienzi LF, Ubaldi FM. Clinical validity and utility of preconception expanded carrier screening for the management of reproductive genetic risk in IVF and general population. Hum Reprod. 2021 Jun 18;36(7):2050-2061. doi: 10.1093/humrep/deab087. PMID: 34021342.
IVF ÖNCESİ TAŞIYICI TARAMA TESTLERİ
Avrupa ve diğer gelişmiş ülkelerdeki tanımıyla nadir hastalıklar; “2000'de 1 ya da daha az sıklıkta görülen, çoğu ilerleyici, metabolik, kronik ve bazıları ölümcül olabilen hastalıklardır”. Dünyada bilinen 8000 civarı nadir hastalık bulunmakta olup her hafta literatürde ortalama 5 yeni hastalık tanımlanmaktadır. Türkiye’de nadir hastalık teşhisi konulan 7 milyon kişi dünyada ise 300 milyon kişi bulunmaktadır. Öte yandan, nadir hastalığı olan çocukların %30’u beş yaşını görmeden kaybedilmekteyken %35’i bir yaşa kadar yaşamaktadır1. Bu hastalıkların %90'ının bilinen bir tedavisinin bulunmaması hastalar, yakınları ve sağlık sistemleri için ciddi psikososyal ve ekonomik sorunlara yol açmaktadır1.
Pek çok genetik hastalığın tedavisi ne yazık ki bulunmamakta olup bu grup hastalığın tedavisi için de “Yetim” ilaçlar kullanılmaktadır. “Yetim” ilaçlar, tedavi edilmesi amaçlanan ama çok nadir hastalıklar olduklarından küçük pazar payı sebebiyle sponsorların ürünün araştırma ve geliştirilmesine yatırdıkları sermayeyi amorti etmelerine izin vermeyeceği için normal pazarlama koşullarında sponsorların geliştirme konusunda isteksiz olduğu ilaçlardır. Nadir hastalıkların teşhis süreci hastalar ve aileleri için olduğu kadar sağlık sistemleri için de büyük bir sorundur. Yakın zamanda yapılmış bir çalışmaya göre hasta başına yapılan teşhis masrafları ortalama 3.692 ABD Doları düzeyindeyken, her başarılı teşhisin maliyeti ise ortalama 21.099 ABD doları düzeyindedir. Bu teşhis zorluklarının yanı sıra, nadir hastalıklarının büyük kısmının bilinen bir tedavisinin olmaması nedeniyle bir hastanın teşhis almış olması, tedavi görebileceği ve iyileşebileceği anlamına gelmez. Tedavisi olan 24 nadir hastalığın ekonomik yükü üzerine yapılan yakın tarihli bir çalışmaya göre de hasta başı yıllık maliyet ortalama 266.000 ABD Dolarıdır2.
Gebelik öncesi genetik tarama, gelecek nesillerin, çoğunun bilinen bir tedavisi bulunmayan nadir hastalıklardan korunmasının önemli bir bileşenidir. Nadir hastalıklı doğumların azaltılması ve nihayetinde eradikasyonu amaçlı olarak tarama programları uygulanmakta olup “önleyici” nitelikte evlilik öncesi, gebelik öncesi ve gebelik sırasında yapılan tarama testleri ile “erken teşhis ve tedaviye imkân sağlayıcı” nitelikte yeni doğan tarama testleri yapılmaktadır.
Avrupa İnsan Genetiği Derneği (European Society of Human Genetics -ESHG) kişilerin önündeki alternatif üreme seçeneklerini değerlendirmeye imkân sağlayacağından, prekonsepsiyonel dönemin taşıyıcılık tarama testleri için en ideal dönem olduğunu belirtmektedir3. Ek olarak Amerikan Obstetrisyenler ve Jinekologlar Derneği’nin (ACOG) 2017 yönergesine göre hamile kalmayı düşünen veya zaten hamile olan tüm kadınlara taşıyıcılık taraması hakkında bilgi verilmesi önerilmektedir. Aynı yönergede prekonsepsiyonel dönemde test yaptırmanın daha fazla seçenek sunacağı ve karar vermek için daha fazla zaman sağlayacağı belirtilmiştir4. Taşıyıcı çiftler için aşağıdaki üreme seçenekleri sunulmuş olup İleri jenerasyonlarda hastalık prevalansının azaltılması/eradikasyonunda bu yaklaşım etkin bir rol oynadığı bildirilmektedir.
- PGT
- Prenatal Tanı
- Donor sperm ya da oosit kullanımı
- Riski kabul etme
- Partner değiştirme
Örneğin akrabalık oranının %60’larda olduğu Suudi Arabistan’da genişletilmiş taşıyıcılık taramasını yaptırmayan çiftler Preimplantasyon Genetik Test (PGT) geri ödemesi kapsamından yararlanamamakta. Yine İsrail hükümeti geniş tarama panellerini 1978 yılında halk sağlığı taramaları kapsamında uygulamaya başlanmış, sonraki aşamalarda bulunan hastalık/gen sıklığına bağlı olarak panel içeriğini popülasyona özgü revize etmiştir. Aslında toplumda hastalıkların eradikasyonu konusuna odaklanmış ülkelerin sağlık politikaları kapsamında tarama testi yaparak genetik hastalık taşıyıcılarını belirlemek ve sağlıklı üreme seçeneklerini uygulamak yer almaktadır.
Tarama programları kapsamında uygulanan yenidoğan taraması biyokimyasal testlerle yapılmakta olup patolojik sonuç varlığında tanı ve tedavinin planlanması için yine biyokimyasal testler kullanılmaktadır. Kimi zaman bu testlerin sonuçları arasında tutarsızlıklar olup semikantitatif olan bu değerlerin doğrulanması ve kesin tanı için moleküler testlerin yapılması gerekmektedir. Son yıllarda, genetik analiz için gereken maliyet ve sürenin sürekli azalmasıyla sonuçlanan yüksek verimli dizileme teknolojilerinde kayda değer bir ilerlemeye sebep olmuş ve yeni doğan taramasında yeni nesil dizilemenin (NGS) daha yaygın bir şekilde kullanılmasını sağlamıştır5.
Mümkün olan en yüksek sağlık standardına ulaşma hakkı olarak tanımlanan “sağlık hakkı” temel insan hakkı olan “yaşama hakkı”nın ayrılmaz unsurudur. Sağlık hakkı; uluslararası insan hakları belgeleri, anayasa ve yasalar ile düzenlenmektedir. Bu kapsamda sağlıklı nesillere ulaşmak ve özellikle tedavisi mümkün olmayan genetik hastalıkların önlenmesine yönelik yapılacak çalışmalar daha büyük önem kazanmaktadır.
Ülkemizde Sağlık Bakanlığı tarafından 24 Ekim 2002'de Kalıtsal Kan Hastalıkları Yönetmeliği'nin yayınlanmasının ardından süreç içinde Bakanlığın belirlediği 41 ilde Talaseminin de içinde bulunduğu kalıtsal kan hastalıklarını önleyebilmek için "Hemoglobinopati Kontrol Programı" başlatılmıştır. Program, hükümetin açıkladığı "100 Günlük Eylem Planı" kapsamında, artık 81 ilde "Evlilik Öncesi Hemoglobinopati Tarama Programı" olarak aile hekimleri tarafından uygulanmaktadır. Talasemi hastalığının dünya üzerinde görülme sıklığı 1/10bin iken ülkemizde 1/6bin olarak bildirilmiştir. Yine SMA hastalığının da ülkemizde yaygın görülen (Dünyada 1/10bin – Türkiye 1/6bin) bir hastalık olması sebebiyle Sağlık Bakanlığı 2021 yılı aralık ayı sonu itibari ile 81 ilde SMA Taşıyıcıcılık Taramasını evlilik öncesi sağlık raporu almak için başvuran çiftler ve halen evli olan çiftlerden de talep edenler için uygulamaya başlamıştır. Mevcut uygulamada öncelikle erkek eşten örnek alınmakta ve şüpheli pozitif bir bulgusu olmadığı sürece kadın eşe test yapılmamaktadır6.
Taşıyıcılık teslerinin sonuçlarını etkileyen genomun zorlu gen bölgeleri nelerdir?
Genom üzerinde ortaya çıkan farklı mutasyon türlerinin belirlenmesi amacıyla farklı moleküler yöntemler kullanılmaktadır. Kistik Fibrozis hastalığından sorumlu CFTR geninde genellikle nokta mutasyonlar görülmekte olup dizileme yöntemleri bu tip mutasyonları etkin bir biçimde taramaktadır. Ancak DMD, SMA gibi hastalıklarda delesyon/duplikasyon tipi mutasyonlar yaygın olarak görülmekte olup ancak MLPA (Multiplex ligation-dependent probe amplification) gibi moleküler yöntemlerle bu tip mutasyonlar tespit edilebilmektedir. Genişletilmiş taşıyıcılık panellerinde çok sayıda gen üzerindeki mutasyonları farklı metotlarla tespit etmeye çalışmak laboratuvara ciddi bir iş yükü getirdiği gibi maliyet açısından da testin yaygınlaşmasını sağlayacak etkin yaklaşımı sınırlandırmaktadır. Günümüz teknolojisinde tek bir test ile farklı genlerdeki farklı mutasyon türlerini tespit etmek araştırmacılar tarafından benimsenen bir hedef haline gelmiştir. NGS bu kapsamda en önemli aday test olup biyoinformatik yaklaşımlarla da bu zorlu bölgeleri tespit etme algoritmaları geliştirilmeye devam etmektedir.
Spinal Muscular Atrofi ve Zorlu SMN1 Geni
SMN1 geninin her iki kopyasında defekt olması SMA hastalığına yol açar. SMA hastalığına yol açan mutasyonların çoğu delesyon ya da gen konversiyonlarıdır. SMA hastalığı taşıyıcısı çiftleri tespit etmede bazı komplikasyonlar bulunmaktadır. 5. Kromozom üzerinde yer alan SMN1 ve SMN2 genleri neredeyse tamamen aynı ikişer kopyaya sahip olup iki gen arasındaki başlıca fark ekzon 7 bölgesinin başında yer alan tek nükleotiddir. SMA hastalığı olan bireylerin %98’inde her iki aleldeki SMN1 geninde de anormallik vardır. Bu anormallik homozigot exon 7 delesyonundan (olguların %95’inde) ya da SMN1 genindeki ekzon 7 delesyonu için birleşik heterozigotluktan veya SMN1 geninde delesyon analizleri ile tespit edilemeyen, dizi analizleri ile tespit edilebilen nokta mutasyonlardan kaynaklanır (Şekil 1). Dolayısıyla SMA hastalığı taşıyıcılığı tek bir moleküler test yöntemi ile tespit edilemeyebilmektedir. Taşıyıcılık tanısındaki bir diğer zorluk ise sessiz mutasyonlardır. Bazı kişilerde bir kromozomda iki SMN1 kopyası varken diğer kromozomda SMN1 gen delesyonunun bulunması sessiz mutasyona neden olmaktadır (2+0 SMA taşıyıcılığı durumu). Standart SMA tanı metotları ile tespit edilemeyen sessiz mutasyonları belirlemek taşıyıcılık taramasında hastalığı tam anlamıyla ekarte edebilmek için oldukça önemlidir. Araştırmacılar sessiz mutasyonu tespit edebilmek amacıyla bu mutasyonla birlikte görülen g.27134T>G mutasyonunu taramaktadırlar. Ashkenazi Yahudileri ve Asya toplumları için ilişkili olan bu varyantın diğer etnik popülasyonlardaki tanı etkinliği azalabilmektedir. Bu nedenle sessiz SMA mutasyonları tarama testleri için hala önemli bir test limitasyonudur.
Bir diğer zorluk ise SMA hastalarının %2’si de novo mutasyon taşımakta olup bu mutasyon ebeveynlerinden kalıtılmamakta ve hasta olguda ilk kez ortaya çıkmaktadır7.
Tüm bu sebeplerden ötürü özellikle üreme seçenekleri hakkında danışmanlık almak isteyen çiftlerde SMA taşıyıcılık test sonuçları değerlendirilirken olası tüm tanı limitasyonları göz önünde bulundurulmalı ve gerekirse ek moleküler testlerle riskler azaltılmalıdır.
Şekil 1 : (A) Normal – her iki kromozomda 2 kopya SMN1 ve 2 kopya SMN2 (B) SMA taşıyıcısı – bir kromozomda 1 kopya SMN1, diğer kromozomda SMN1 gen kaybı (C) sessiz SMA taşıyıcısı – bir kromozomda SMN1 gen duplikasyonu, diğer kromozomda SMN1 gen kaybı (D) Taşıyıcı - Kromozomun birinde normal SMN1 kopyası, diğer kromozomda SMN1 nokta mutasyonu
Konjenital Adrenal Hiperplazi ve Zorlu CYP21A2 Geni:
Konjenital Adrenal Hiperplazi bir grup resesif hastalık olup 21-Hidroksilaz Eksikliği (21-OHD) en yaygın formudur. Olguların %90-95’inde görülen 21-OHD, CYP21A2 genindeki mutasyonlar nedeniyle ortaya çıkmaktadır. Geniş taşıyıcılık testlerinde gen panelleri içerisinde mutlaka bulunmakla birlikte gen ve psödogeni (CYP21A2 aktif gen, CYP21P psödogen) arasında çok yüksek homoloji olduğundan moleküler tanısı oldukça zorludur. DNA dizisinin intergenik rekombinasyonları sonucunda delesyonlar, duplikasyonlar, gen konversiyonları ve nokta mutasyonlar meydana gelir. CYP21A2’nin psödogeninin bulunması nedeniyle 21-hidroksilaz enzim eksikliğine bağlı gelişen KAH’a sadece CYP21A2 genine yönelik dizi analizi çalışmalarıyla doğru tanı konulmasında çoğu kez aksaklıklar yaşanmaktadır. Ayrıca dizi analizinde saptanması mümkün olmayan kopya sayısı değişikliklerinin belirlenmesi için ek bazı test metodolojilerine de (MLPA) ihtiyaç duyulmaktadır8.
Alfa Talasemi ve Zorlu HBA Geni
Alfa Talasemi, α globulin zincir geninin bir tanesinden dört taneye kadar hepsinin delesyonu veya nokta mutasyonu sonucu, delesyona uğrayan gen miktarına bağlı olarak sessiz anemiden ölümcül hastalık formuna kadar farklı klinik özelliklerdeki bir hastalıktır. α1-globini kodlayan HBA1 ve α2-globini kodlayan HBA2 genleri 16. kromozomun kısa kolunun uç bölgesinde (16p13.3) yerleşik üçer ekzon ve ikişer introndan oluşmaktadır. Etkilenmiş hastaların %90’ında delesyon, %10’unda ise nokta mutasyonları gözlenir. Bu nedenle geniş NGS tabanlı tarama testlerinde bu delesyon tipi mutasyonları tespit etmek oldukça güçtür. (9)
Gaucher Hastalığı ve Zorlu GBA Geni :
Parkinson hastalığına neden olan başlıca risk faktörlerden biri olan Gaucher Hastalığı GBA geninde görülen mutasyonlar sonucu ortaya çıkmaktadır. Son zamanlarda GBA geninin moleküler analizi için tek gen ya da panellerde NGS metodu ile kullanılmaktadır. Ancak Yüksek homolojideki GBAP1 psödogeninin varlığı nedeniyle ortaya çıkan kompleks gen-psödogen etkileşimleri nedeniyle NGS analizi ile mutasyon tespitinde zorluklar oluşmaktadır. Bu nedenle hatalı tanıyı önlemek için dizileme ve analiz yaklaşımlarında bazı stratejiler oluşturulmalıdır. (10)
Kistik Fibrozis ve Zorlu CFTR Geni :
Kistik Fibrozis hastalığı etnik panellerden panetnik panellere taşıyıcılık taramalarındaki en önemli hedef genlerin başında gelmektedir. Her ne kadar CFTR gen mutasyonları çok detaylı incelenmiş ve literatürde birçok çalışma yer almış olsa da her geçen gün özellikle intronik/derin intronik bölgelerde yeni mutasyonlar tanımlanmakta. Sadece kodlayıcı (ekzon) bölgelerin hedeflendiği dizilemelerde bu kodlayıcı olmayan bölge (intron) mutasyonları atlanabilmektedir. Ayrıca delesyon/duplikasyon tipi mutasyonların da varlığı (%3-5) MLPA gibi ek moleküler testlerin yapılmasını gerektirmektedir. Tüm bu mutasyonları tek bir NGS metodu ile yapabilmek taşıyıcılık test panellerinin yaygın ve uygun maliyetli kullanımı için önemli bir hedef haline gelmiştir. (11)
Taşıyıcılık Tarama Programının Tarihçesi:
Genetik taşıyıcılık testleri uzun yıllardır hasta çocuk sahibi olma riski olan genetik hastalık taşıyıcılarının tespit edilebilmesi, bu hastalıkların insidanslarının azaltılması ve taşıyıcı çiftlerin sağlıklı çocuk sahibi olmalarının sağlanmasında önemli bir rol oynamaktadır. Bu tip testlerin kullanımı 1970’lerde Ashkenazi Yahudi popülasyonlarında Tay Sacs hastalığını taramak için kullanılırken 1980’lerde akdeniz ülkelerinde Beta Talasemi ve Orak Hücre Anemisi toplum taramaları başlatıldı. 1993’te aile öyküsü ve etnik kökenine bakılmaksızın Kistik Fibrozis hastalığının toplum taraması programına alınması “Evrensel Taşıyıcılık Testi” tabirini gündeme getirdi. 2010 yılında Consyl isimli biyoteknoloji firması 100 mendeliyen kalıtılan geni içeren bir tarama testini bildirirken 2011 de Bell ve arkadaşları 437 genin tarandığı panelde “Yeni Nesil Dizileme” (Next Generation Sequencing – NGS) teknolojisini kullanan ilk grup olmuştur12. O günden sonra çok hızlı bir biçimde pek çok taşıyıcılık tarama paneli servis edilmiş olup global bir çözüm olarak tüm ekzom / tüm genom tabanlı testlerin kullanımı artmıştır.
Geniş Tarama Testleri (Expanded Carrier Screening - ECS) ilk uygulamaya girdiği dönmelerde “Hot Spot” olarak tabir edilen tüm gen taramaları yerine ilgili genlerdeki yaygın mutasyonları tarayan sınırlı tarama panelleri olarak gündeme gelmişti. Uzun yıllar bu yaklaşımla tarama testleri yapılmış olmakla beraber dizileme teknolojilerindeki gelişmeler ve test maliyetlerindeki iyileşme tüm gen taramalarına imkân verir hale gelmiştir. Bu sayede, intronik ve derin intronik bölgeler gibi genomun patojenik varyant taşıma riski görece daha düşük bölgelerinde de mutasyon analizleri yapılabilir hale gelmiş hastaların nadir taşıyabilecekleri bozukluklar da ekarte edilebilmiştir.
Bu hızlı ve düşük maliyetli testlerle (pan-etnik ya da evrensel) etnik kökene bakılmaksızın eşitlik çerçevesinde ve kişileri damgalamadan taşıyıcılıklarını belirleme şansı kazanılmıştır.
Önceden “Amerikan Kadın Hastalıkları ve Doğum Uzmanları Cemiyeti” (American College of Obstetricians and Gynecologists-ACOG) taşıyıcılık testlerinin etnisiteye göre dizayn edilmesini önermekteydi ve belli hastalıklar için artmış riski olan popülasyonlara odaklanılmaktaydı13.
ACOG 2011 yılında yayınladığı komite kararlarında; taşıyıcılık testlerinin etnik-spesifik, Pan-etnik ve genişletilmiş taşıyıcılık panelleri olarak servis edilebileceğini, evrensel Kistik Fibrozis taramasına SMA ve hemoglobinopati taramasının da eklenmesini önermiştir14. SMA’yı da içeren genetik kondisyonların etnik gruplarla sınırlı olmadığı, globalleşen dünyada etnik grupların karıştığı belirtilmiştir. Bu kapsamda prekonsepsiyonel taramalarda genişletilmiş tarama testlerinin yapılmasının doğru bir strateji olduğu belirtilmiştir. Ancak Kıbrıs, Sardinya Adası gibi Talasemi hastalık insidansı yüksek populasyonlar ve Ashkenazi Yahudi toplumlar dışında taşıyıcılık taramalarının halk sağlığı taramaları kapsamında gebelik planlayan tüm popülasyona uygulanmasının bazı sakıncaları vardır.
- Psikolojik etkiler
- Damgalamak/ayrımcılık
- Kişisel seçimleri usulsüzce baskılaması ve kişisel seçim özgürlüğünü sınırlaması
- Aile kurmak isteyen çiftler arasında seçim kriteri haline gelebilme durumu
Taşıyıcı bireylerin tespitine yönelik testler iki ayrı grupta toplanabilir:
Taşıyıcılık Taraması: Belli bir hastalık için hasta çocuk sahibi olmada artmış riski bulunmayan çiftler
Taşıyıcılık Testi: Eş ya da aile öykülerinde pozitif bulgusu olan hasta çocuk sahibi olma riski artmış çiftler
Varyant Sınıflandırma American College of Medical Genetics – 2010 Genom dizilemesi ile bir bireyde 4 milyon civarında varyant tespit edilirken, genomun protein kodlayan ve %1’lik kısmını oluşturan ekzom dizilemesi sonucunda yaklaşık 20,000 varyant elde edilmektedir. Bu kadar varyanttan hastalık etmeni olan mutasyonu bulmak samanlıkta iğne aramaya benzemektedir. Bu noktada ailenin detaylı klinik bilgisine sahip olmak çok önemlidir (Lohmann ve ark.). Etkin bir biyoinformatik filtreleme için temelde varyantın popülasyon frekansı düşük olanların seçilmesi ile varyant sayısı azaltılabilir ve sonrasında kalıtım paterni ve fenotipik bulguların eklenmesi ile hastalık etmeni mutasyona ulaşılabilir. Klinik tanı net olmadığında kullanılan tüm genom ve ekzom testleri pek çok hasta için yüz güldürücü sonuçlar vermekle birlikte bu yüksek çıktılı engin genom datasının bizlere sunduğu, literatürde daha önceden hiç bildirilmemiş yeni varyantlar şüpheli raporların üretilmesine de neden olmaktadır. Buradaki tehlikeyi anlamak için yeni jenerasyon mutasyon sınıflandırması hakkında fikir sahibi olmamız gerektiğinden dünya üzerinde pek çok klinik raporun dayanağı olan American College of Medical Genetics and Genomics (ACMG) kılavuzu kriterlerine göre mutasyon skorlamasından bahsetmemiz gerekmektedir15 (Şekil 2).
Şekil 2 : American College of Medical Genetics and Genomics (ACMG) kılavuzu kriterlerine göre mutasyon skorlaması
Taşıyıcılık Tarama Testlerinde Hangi Genlere Bakılmaktadır?
Taşıyıcılık taraması erken dönem ölümle seyreden ya da engelli hasta çocuk sahibi olma riski olan taşıyıcı bireylerin tespit edilmesini sağlayan genetik testlerin bütünüdür. Bu testler genellikle resesif ve X’e bağlı aktarılan hastalıkları taramaktadır. Pozitif aile öyküsü olan erişkinlere taşıyıcılık taraması önermek pek çok ülkede standart bir uygulama olmasına rağmen bu yolla taşıyıcı çiftlerin çok az bir kısmı tespit edilebilmektedir. Pek çok hasta çocuk aile öyküsünde hasta birey olmaksızın doğmakta ve sadece yüksek risk kategorisindeki akraba evliliği yapan çiftler tarama testi talep etmektedirler.
Amerikan Tıbbi Genetik ve Genomik Cemiyeti (American College of Medical Genetics and Genomics -ACMG) Ashkenazi Yahudilerin taranması için ilkin 9 gen içeren bir pan-etnik panelin kullanılmasını önermiştir.3 ACOG 2017 yılında yayınladığı komite raporunda 14 genin taranmasını tavsiye etmiştir4. Genel olarak SMA, Tay Sacs, Beta Talasemi, Kistik Fibrozis, Frajil X hastalıklarının tarandığı etnik paneller yaygın olarak kullanılmaktayken dizileme teknolojilerindeki gelişmeler ve test maliyetlerindeki azalma 100-600 gen içerikli panetnik panellerin de yaygınlaşmasına neden olmuştur. Ancak bu testlerde gen panellerinin içeriğinin belirlenmesi hala tartışma konusudur. Üreme tıbbında kullanılmak üzere kişinin etnik kökeni ve aile geçmişi bilgisine gerek duyulmaksızın uygulanabilecek panetnik ve yüksek klinik fayda sağlayan taşıyıcılık testlerinin kullanımı her geçen artmaktadır. Her toplumun kendi epidemiyolojisine uyumlu, farklı ECS yaklaşımlarına ihtiyacı vardır.
Popülasyon genetik çeşitliliği, çevresel, toplumsal faktörler ve hayatta kalma oranları nedeniyle coğrafi bölgeler arasında sıklık bakımından değişiklik gösteren 1800'den fazla kalıtsal nadir hastalık olduğu tahmin edilmektedir16. Avrupa'da sadece 12 nadir hastalığın tüm vakaların %90'ını oluşturduğu bilinmektedir. Bununla birlikte Türkiye ve Orta Doğu ülkelerinde aynı 12 hastalığın gözlenme oranı %35'tir17. Dolayısıyla ülkemizde dünyada sık görülen hastalıklara nazaran oldukça nadir görülen genetik hastalıklar daha yaygındır. Bu durumda evlilik öncesi tarama programlarını genişletmek ve ülkenin epidemiyolojisine uyumlu daha fazla hastalığı tarayan programlara geçmek bu hastalıkların eradikasyonunda büyük önem taşımaktadır. Akraba evliliği oranlarının yüksek olduğu toplumlarda, nadir heterozigot (asemptomatik) taşıyıcılıkların, çocuklarda homozigot olarak (hasta) ortaya çıkma olasılığı artmakta ve bebeklerin bilinen nadir hastalıklardan veya yeni sendromlardan etkilenme riski yükselmektedir.
Göç dalgaları kaynaklı gen havuzumuzun değişmesi ile birlikte yeni genetik hastalıklar ortaya çıkabilmektedir. Özellikle ülkemizde göç eden ülkelerin de akraba evliliği oranının yüksek olduğu göz önünde bulundurulursa genetik hastalıklı çocuk doğumlarının artışı önemli bir sağlık problemini de beraberinde getirmektedir. Benzer şekilde Türkiye'de 3,6 milyon kayıtlı Suriyeli, diğer milletlerden (Irak, Afgan, diğerleri) de 320.000 mülteci yaşamaktadır18. Türkiye İstatistik Kurumu, Suriyeli mültecilerin %30'unun üreme çağında olduğunu, evliliklerin yaklaşık %40'ının akraba evliliği olduğunu ve son dört yılda 518.730 doğum gerçekleştiğini bildirmektedir. Sürekli göç, mülteci topluluklarındaki yüksek üreme ve akraba evliliği oranları nedeniyle, nadir hastalıklarla mücadele Türkiye için öncelikli bir halk sağlığı problemi olarak yeni bir boyut kazanmıştır. Yaygın nadir hastalıkların henüz bilinmeyen genetik varyantları ve yeni sendromlar Türkiye ve Avrupa'nın genetik yapısına dahil olmakta ve kısa vadede bunların görülme sıklıklarının artması beklenmektedir.
ECS, sağlıklı ebeveyn adaylarına gebelik öncesi genetik hastalık taşıyıcılık risklerinin tespit edebilmek amacıyla yapılan, klasik genetik taramaların aksine, yüzlerce hedef gen bölgesinin tek bir testte taranmasını sağlayan, uygulanmasında kişilerin etnik ya da sağlık durumuyla ilgili herhangi bir kriter aranmayan bir genetik taramadır. Bu taramanın yapılması ve nadir hastalık taşıyıcılık durumlarının raporlanması, ebeveyn adaylarının üreme seçenekleri hakkında bilgilendirilmesi ve sağlıklı bebeklerin doğumu için çok önemlidir.
TÜİK aile yapısı araştırması 2016 verilerine göre; Türkiye’de ortalama akraba evliliği oranı %20.9 olup Diyarbakır’da %37.2 Konya’da %23.3’tür. Akraba evliliklerinde çiftler özellikle nadir resesif hastalıkları taşıdıklarından bu hastalıkların pek çoğu genişletilmiş tarama panelleri kapsamında yer almadığından bu tür ticari rutin testler akraba evliliği yapan çiftler ya da akraba evliliğinin yüksek oranda görüldüğü ülkelerin tarama programları için yeterli gelmemektedir. Şimdilerde tarama programları içine girmeye başlayan tüm ekzom (Whole Exome Sequencing-WES) ve tüm genom (Whole Genome Sequencing-WGS) dizilemeleri genetik olarak izole popülasyonların nadir hastalıklarını da içeren tüm hastalıkların taranmasına imkân sağlayacaktır. Ancak yeni nesil dizileme teknolojileri ile elde edilen bu büyük varyant datasının en tehlikeli çıktısı olan bildirilmemiş değişimlerin yorumlanması bu testlerin sonuçlarını kompleks hale getirmektedir. Klinik fenotipe etkisi bilinmeyen, “VUS- Variant of Unsignificance” olarak tabir edilen bu varyantlar patojenik ya da beningn olabilirler. ClinVar gibi halka açık veri tabanlarında varyantların laboratuvarlar arası patojenite değerlendirmeleri arasındaki farklılıklar da sonuçların yorumlanmasında zorlukları beraberinde getirmekte. Bir laboratuvarın patojenik dediği bir varyanta başka bir laboratuvar VUS diyebiliyor. Bunların ötesinde farklı laboratuvarlar aynı kişi için birbirinden tamamen farklı sonuçlar verebiliyor. Genomik alanda teknoloji hızla ilerlerken verileri yorumlamada aynı hızı gösterememekte olduğumuzdan patojenik etkisi net olmayan varyantların genetik danışmanlığının verilmesinde detaylı hasta bilgilendirmesinin yapılması ve hasta onamı ile prenatal ve preimplantasyon testlerin gerçekleştirilmesi büyük önem taşımaktadır.
Panel içeriği genişledikçe bu testlerde duyarlılık sorunları nedeniyle klinik fayda ve uygulanabilirlik yönünden iyileştirmelere ihtiyaç bulunmaktadır. Buna ek olarak bir ECS panel içeriğinin ne olması gerektiği konusunda bir fikir birliği bulunmamaktadır. Bazı ECS testleri yukarıda belirtilen sorunu panele dahil edilen gen sayısının arttırılması ile çözmeye çalışmaktadır. Ancak hem kişisel sağlık hem de halk sağlığı açısından, panelde yer alan gen sayısının artırılması, çiftler arasındaki kümülatif mutasyonların tespiti üzerinde sınırlı bir etkiye sahiptir yani tespit edilen mutasyon sayısı, paneldeki gen sayısı arttıkça iyileşmemektedir. Bu durum panel gen listesi kürasyonunun öneminin altını çizmektedir.
Günümüzde yaygın görüş özgüllük, duyarlılık ve dengeli okuma derinliğinin panelde yer alan gen sayısından daha yüksek önem taşıdığı, 100 hastalığı %100 duyarlılıkla tarayan bir taşıyıcı testinin, 1000 hastalığı %10 duyarlılıkla tarayan tarama testinden daha etkili olduğu yönündedir20.
Genetik veri tabanları, nadir hastalıkların varyantları ve bunların frekansları hakkında bilgi sağlamaktadır, ancak belirli etnik gruplar, yaş grupları ve cinsiyetlere dair veriler yetersiz kalmaktadır. Bu nedenle, 1.000'den fazla monogenik nadir hastalıktan hangilerinin, hangi genlerin ve bunlarla ilişkili hangi varyantların bir ECS paneline dahil edilmesi gerektiği hala önemli bir tartışma konusudur. ECS panelleri, varyantların tespitinde farklı teknik zorluklar barındıran hem kodlayan hem de kodlamayan bölgeleri kapsayan bir gen koleksiyonu içermektedir. Gen panelinin tasarımında, hangi genlerin dahil edileceğine karar verilirken, hedef gen bölgelerini zenginleştirme yönteminin seçimi, okuma derinliğinin belirlenmesi ve panele özgü analiz yazılımı adaptasyonu gibi özellikler açısından kapsayıcı ama aynı anda da hedefe yönelik ve odaklı olmak gerekmektedir21. Genomdaki karmaşık bölgelerdeki varyantların saptanmasında birden fazla akış hattı ve analiz yöntemi kullanılması gerekmektedir. Teknik olarak zorlayıcı varyant türleri için hem algoritma tasarımı hem de yeni verilerle model geliştirilmesi yaklaşımı ile, yapay zekâ tabanlı biyoinformatik araçların kullanımı oldukça önemli bir hale gelmiştir.
Taşıyıcılık testlerinde temel hedef, baştan sona tüm süreci kapsayan bir NGS testi sunmak ve hedef listesindeki DMD, SMA, CFTR gibi zorlu bölgeler için ek test gereksinimini ortadan kaldırmaktır. Klinik düzeyde duyarlılığın sağlanamadığı zorlu genom bölgeleri (psödogenler, gen konversiyonları vb) ve kopya sayısı değişiklikleri (Copy Number Variation – CNV) tespiti için ek bazı mutasyon türüne özgü moleküler test metotları kullanma ihtimali bulunmaktadır.
Varyant sınıflandırması statik bir olgu değildir; Bir varyantın sınıf I, II ya da III olarak sınıflandırılması, kullanılan veri tabanında biriken genetik veriye göre değişebilir. Ayrıca bir varyantın sınıflandırılması farklı veri tabanları arasında farklılık gösterebilir ve bu durum özellikle VUS'ların raporlanmasında sorun oluşturabilir. VUS'ların doğru ve klinik faydayı arttıracak şekilde raporlanması rezidüel (tarama sonuçları herhangi bir taşıyıcılık işaret etmediği durumlarda bile ebeveynlerin taşıyıcı olma olasılığı) riskin azaltılması için büyük önem taşımakta bununla birlikte bu sorumluluk mevcut testler tarafından göz ardı edilerek klinisyene bırakılmaktadır.
Mevcut ECS analizlerinde bir yaklaşım yalnızca patojenik/muhtemel patojenik varyantları (sınıf I ve II) raporlayarak VUS'lar dikkate alınmamakta ve bu da risk altındaki çiftlerin gözden kaçmasına neden olabilmektedir. Eşlerden birinin sınıf I/II varyant taşıyıcısı ve diğerinin ise kodlayan VUS taşıyıcısı olduğu çiftlerin %20'sinin, frekans verilerinin birikimine bağlı olarak, her ikisi de sınıf I/II taşıyıcısı olacak şekilde yeniden sınıflandırılacağı tahmin edilmektedir. Mevcut VUS anlayışı, ECS'de VUS raporlamasını engellese de, bu bulgular VUS'ların sınıflandırmasının ve raporlanmasının önemini vurgulamaktadır. Klinisyene hastalarına doğru üreme tercihleri sunabilmesini sağlayacak, filtrelenmiş VUS bilgisini de içeren bir tarama raporu sağlayabilmek için bulguların gözden geçirilmesi ve karanlıkta kalan noktaların en aza indirilmesi büyük önem taşımaktadır.
ESHG - Genetik tarama programlarının prensipleri, teknikler, uygulamalar, politikalar3.
- Genetik tarama amaçları ve hedef popülasyon iyi tanımlanmalı
- Laboratuvar kalite kontrolleri sıkı olmalı
- Sonuçların limitleri açıkça belirtilmeli
- Otoriteler kişilerin taşıyıcılık durumlarına ait gizliliği sağlayabilmeli ve kişisel genomik verilerin saklanması ve korunmasını güvence altına almalı
- Kişi ve aile mahremiyetini sağlayabilmeli
- Uzun vadedeki sonuçlar monitörize edilebilmeli
- Eğitim programları düzenlenmeli ve genetik danışmanlık verilmeli
Taşıyıcılık testleri kişilerin üreme kararlarının daha güvenilir olmasını sağlamaktadır. Günümüzde geniş taşıyıcılık tarama panellerinin maliyet etkinliği arttığından özellikle IVF uygulamaları öncesi çiftlerin genetik taşıyıcılık risklerinin belirlenmesinde önemli bir uygulama haline gelmektedir. 3877 olgunun dahil edildiği IVF öncesi yapılan ECS test bulgularına göre olguların %10.4’ü en az 1 gende patojenik ya da olası patojenik varyant taşımaktadırlar. Bu çalımada incelenen 766 çiftin %22.6’sında tek eş mutasyon taşıyıcılığı bulunmuşken %2.6’sı risk altındaki her iki eş taşıyıcıları olarak tespit edilmiştir22. Bu kapsamda IVF öncesi taşıyıcılık panellerinin kullanımı hasta çocuk doğumlarının önlenmesinde önemli bir uygulama haline gelmeye başlamaktadır. Ancak özellikle akraba evliliği yüksek toplumlarda sınırlı gen panellerinin yerine daha geniş taşıyıcılık test panellerinin devreye girmesi amaçlanmaktadır. Bu noktada elde edilen genomik verinin etkin değerlendirmesine imkân sağlayacak olan hastalık yapıcı varyant bilgisinin artması hedeflenmektedir. Preimplantasyon Genetik Testlerin günümüz teknolojisinde başarıyla uygulanıyor olması taşıyıcılık testleri ile risk taşıyan çiftler için önemli bir seçenek oluşturmaktadır. Hastalar için taşıyıcılıktan tanıya bir tespit imkânı veren bu üreme seçeneklerinin kullanımı her geçen gün artmaktadır.
Kaynaklar
- https://globalgenes.org/rare-diseases-facts-statistics/
- Andreu P, Karam J, Child C, Chiesi G, Cioffi G. The Burden of Rare Diseases: An Economic Evaluation. :7.
- Henneman, L., Borry, P., Chokoshvili, D. et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet 24, e1–e12 (2016). https://doi.org/10.1038/ejhg.2015.271
- Committee Opinion. (2017b). Committee Opinion No 691 Summary. Obstetrics & Gynecology, 129(3), 597–599. https://doi.org/10.1097/ aog.0000000000001948
- Remec ZI, Trebusak Podkrajsek K, Repic Lampret B, Kovac J, Groselj U, Tesovnik T, Battelino T, Debeljak M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front Genet. 2021 May 26;12:662254. doi: 10.3389/fgene.2021.662254. PMID: 34122514; PMCID: PMC8188483
- https://hsgm.saglik.gov.tr/tr/cocukergen-tp-
- Wirth B, Schmidt T, Hahnen E, Rudnik-Schöneborn S, Krawczak M, Müller-Myhsok B, Schönling J, Zerres K. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet. 1997 Nov;61(5):1102-11. doi: 10.1086/301608. PMID: 9345102; PMCID: PMC1716038.
- Nan MN, Roig R, Martínez S, Rives J, Urgell E, Espinós JJ, Tirado M, Carreras G, Aulinas A, Webb SM, Corcoy R, Blanco-Vaca F, Tondo M. Comprehensive Genetic Testing of CYP21A2: A Retrospective Analysis in Patients with Suspected Congenital Adrenal Hyperplasia. J Clin Med. 2021 Mar 12;10(6):1183. doi: 10.3390/jcm10061183. PMID: 33809035; PMCID: PMC8001222.ALFA
- Vijian D, Wan Ab Rahman WS, Ponnuraj KT, Zulkafli Z, Mohd Noor NH. Molecular Detection of Alpha Thalassemia: A Review of Prevalent Techniques. Medeni Med J. 2021;36(3):257-269. doi: 10.5222/MMJ.2021.14603. Epub 2021 Sep 30. PMID: 34915685; PMCID: PMC8565582.
- Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J Mol Diagn. 2017 Sep;19(5):733-741. doi: 10.1016/j.jmoldx.2017.05.005. Epub 2017 Jul 18. PMID: 28727984.
- Morris-Rosendahl DJ, Edwards M, McDonnell MJ, John S, Alton EWFW, Davies JC, Simmonds NJ. Whole-Gene Sequencing of CFTR Reveals a High Prevalence of the Intronic Variant c.3874-4522A>G in Cystic Fibrosis. Am J Respir Crit Care Med. 2020 Jun 1;201(11):1438-1441. doi: 10.1164/rccm.201908-1541LE. PMID: 32017858.
- Beaudet AL (2015) Global genetic carrier testing: a vision for the future. Genome Med 7:79
- Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. ACOG Committee Opinion No. 442. American College of Obstetricians and Gynecologists. Obstet Gynecol 2009;114:950–3.
- Committee Opinion No. 486: Update on Carrier Screening for Cystic Fibrosis. Obstetrics & Gynecology: April 2011 - Volume 117 - Issue 4 - p 1028-1031 doi: 10.1097/AOG.0b013e31821922c2
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
- Cornel MC, Rigter T, Jansen ME, Henneman L. Neonatal and carrier screening for rare diseases: how innovation challenges screening criteria worldwide. J Community Genet. 2021;12(2):257-265. doi:1007/s12687-020-00488-y
- Beauchamp KA, Muzzey D, Wong KK, et al. Systematic design and comparison of expanded carrier screening panels. Genetics in Medicine. 2018;20(1):55-63. doi:1038/gim.2017.69Refugees and Asylum Seekers in Turkey – UNHCR Türkiye. Accessed August 11, 2022. https://www.unhcr.org/tr/en/refugees-and-asylum-seekers-in-turkey
- Refugees and Asylum Seekers in Turkey – UNHCR Türkiye. Accessed August 11, 2022. https://www.unhcr.org/tr/en/refugees-and-asylum-seekers-in-turkey
- Ben-Shachar R, Svenson A, Goldberg JD, Muzzey D. A data-driven evaluation of the size and content of expanded carrier screening panels. Genetics in Medicine. 2019;21(9):1931-1939. doi:1038/s41436-019-0466-5
- Hogan GJ, Vysotskaia VS, Beauchamp KA, Seisenberger S, Grauman PV, Haas KR, Hong SH, Jeon D, Kash S, Lai HH, Melroy LM, Theilmann MR, Chu CS, Iori K, Maguire JR, Evans EA, Haque IS, Mar-Heyming R, Kang HP, Muzzey D. Validation of an Expanded Carrier Screen that Optimizes Sensitivity via Full-Exon Sequencing and Panel-wide Copy Number Variant Identification. Clin Chem. 2018 Jul;64(7):1063-1073. doi: 10.1373/clinchem.2018.286823. Epub 2018 May 14. PMID: 29760218.
- Liu Z, Zhu L, Roberts R, Tong W. Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends in Genetics. 2019;35(11):852-867. doi:1016/j.tig.2019.08.006
- Capalbo A, Fabiani M, Caroselli S, Poli M, Girardi L, Patassini C, Favero F, Cimadomo D, Vaiarelli A, Simon C, Rienzi LF, Ubaldi FM. Clinical validity and utility of preconception expanded carrier screening for the management of reproductive genetic risk in IVF and general population. Hum Reprod. 2021 Jun 18;36(7):2050-2061. doi: 10.1093/humrep/deab087. PMID: 34021342.